
动脉粥样硬化是全球心血管疾病的‘头号杀手’,即便现有降脂、抗炎疗法不断升级,仍有大量患者面临心血管事件复发风险。
为什么血脂达标了,心梗风险依然存在?
为什么抗炎治疗不能替代他汀?
为何单一疗法始终无法彻底攻克动脉粥样硬化?
中国医学科学院阜外医院李建军教授团队提出的全新假说,给出了核心答案。
尽管动脉粥样硬化的多维度防治手段取得了诸多进展,但该病仍是全球范围内心血管疾病发病和死亡的首要原因。目前临床治疗的现状仍不尽如人意,这可能源于对其潜在发病机制的理解尚不充分。长期以来,胆固醇假说与炎症假说这两大主流理论一直在动脉粥样硬化的发病机制研究中占据主导地位。
胆固醇假说将低密度脂蛋白胆固醇(LDL-C)认定为动脉粥样硬化斑块发生与进展的核心致病因素,同时残余胆固醇也被证实是导致心血管残余风险的重要致动脉粥样硬化脂质组分。与之相对,炎症假说则将动脉粥样硬化重新定义为一种适应性异常的免疫反应,强调细胞因子信号传导、免疫细胞募集以及慢性血管损伤在疾病发生发展中的作用。越来越多的研究证据表明,胆固醇代谢与炎症反应并非并行的致病因素,而是相互交织的病理过程。但目前的研究大多将二者视为相互联系但概念上可分离的过程,尚无一个理论框架能将其系统地整合统一。

基于此,中国医学科学院阜外医院李建军教授团队于《JACC: Asia》杂志发表文章“Cholesterol-Inflammation Fusion Hypothesis in Atherosclerosis: An Evolving Paradigm in Pathogenesis and Therapy”,提出胆固醇-炎症融合假说。该假说在现有理论基础上进一步发展,明确将动脉粥样硬化定义为一种由胆固醇代谢紊乱与慢性血管炎症形成的功能性不可分割、自我维持的双向融合环路所驱动的疾病。本文将阐述胆固醇-炎症融合假说的核心理论原则、分子机制研究进展、临床验证依据以及治疗转化意义,同时也将指出该假说在转化应用中面临的挑战和未来的研究重点。
动脉粥样硬化胆固醇-炎症融合假说:发病机制及治疗的演进范式
— JACC:Asia —
动脉粥样硬化发病机制的研究历史回顾
胆固醇假说的起源可追溯至1913年一项开创性实验,该研究成功诱导出与人类动脉粥样硬化相似的动脉病变,确立了胆固醇在斑块形成中的核心始动作用,也为后续以脂质调控为核心的干预手段奠定了理论基础。20世纪末,有学者首次明确将动脉粥样硬化定义为一种炎症性疾病,将血管病变重新阐释为血管内皮损伤引发的慢性免疫反应。这一观点在21世纪得到了确凿的临床证据支持,其中最具代表性的是卡那单抗抗炎抗血栓结局研究(CANTOS)。
随着研究的深入,人们逐渐发现,无论是以胆固醇为核心还是以炎症为核心的单一模型,都无法完整阐释动脉粥样硬化的复杂病理机制。即便实现了理想的降脂治疗,心血管残余风险依然存在;而单纯的抗炎策略也无法替代胆固醇调控的作用。分子生物学和单细胞技术的最新研究进展进一步揭示,脂质蓄积与免疫激活并非相互独立的过程,二者深度交织,并共同参与炎症小体激活、胆固醇外流障碍、巨噬细胞表型异常等多条细胞通路的调控。这一认知的演进,为胆固醇-炎症融合假说的提出奠定了基础。
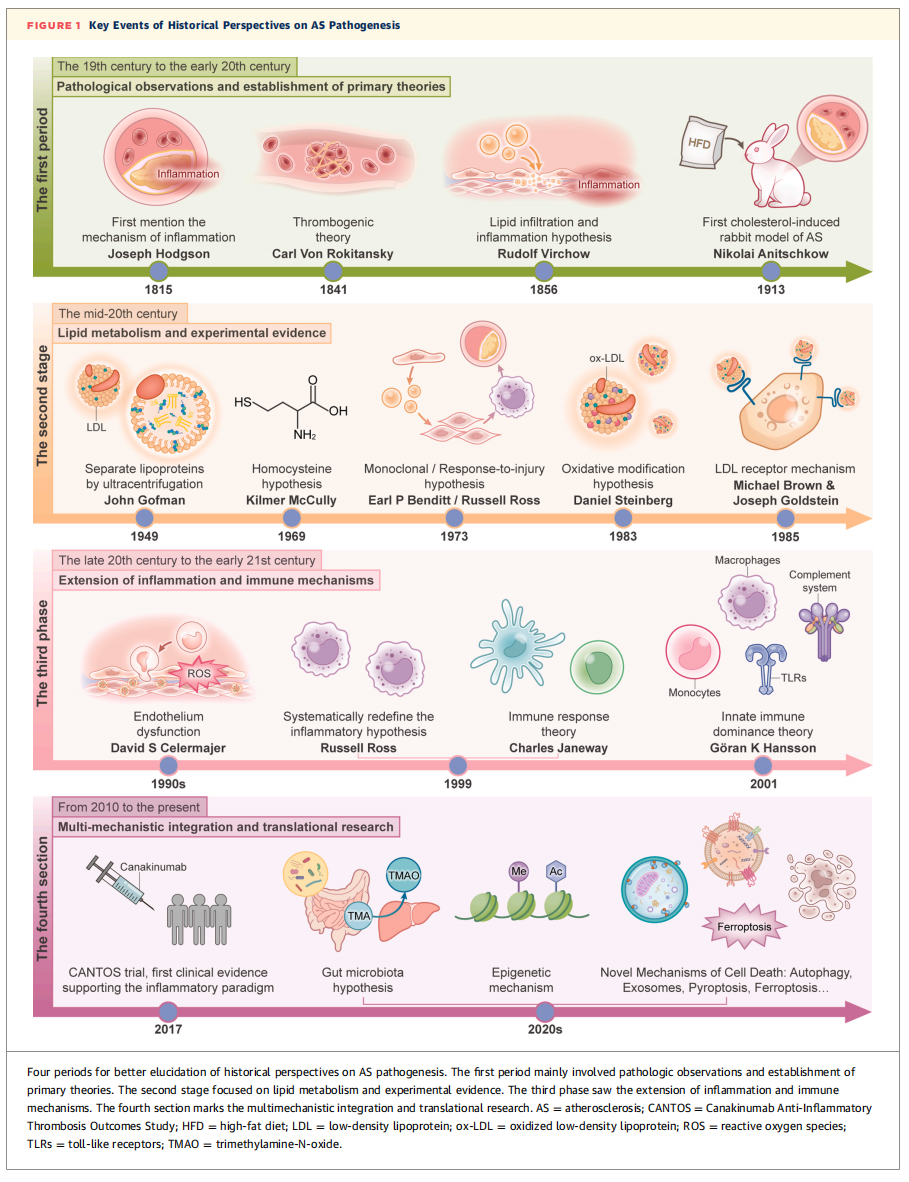
图1 动脉粥样硬化发病机制研究的历史进程关键事件
胆固醇-炎症融合假说:核心概念与基本原则
胆固醇-炎症融合假说将动脉粥样硬化定义为一种由胆固醇代谢紊乱与慢性血管炎症深度交织、不可分割所引发的疾病。实际上,不断涌现的研究证据表明,这两个过程存在相互强化的作用,会形成一个自我维持的恶性循环,推动动脉粥样硬化斑块的形成、进展与失稳。
在该假说的理论框架中(图2A),胆固醇兼具底物与信号分子的双重作用。过量的低密度脂蛋白(LDL)及其修饰形式会在血管内膜蓄积,并作为损伤相关分子模式,促进泡沫细胞形成与炎症信号通路的激活。另一方面,LDL-C酯经溶酶体水解产生的游离胆固醇在细胞内过度蓄积,会析出形成胆固醇结晶,同样可激活机体的固有免疫反应。与之相对,炎症介质会破坏脂质代谢平衡:白介素(IL)-1β、肿瘤坏死因子(TNF)-α等细胞因子会下调腺苷三磷酸结合盒转运体A1和G1的表达,破坏高密度脂蛋白(HDL)介导的胆固醇外流过程,加剧巨噬细胞与血管平滑肌细胞内的脂质滞留,而这两种细胞也是斑块中泡沫细胞形成的重要来源。
总体而言,胆固醇-炎症融合假说的核心基本原则可总结为三点:
1. 双向交互性:胆固醇代谢紊乱会驱动炎症反应的发生,而炎症反应又会进一步损害胆固醇的正常代谢过程;
2. 自我维持性:一旦脂质沉积与免疫激活的循环形成,便会持续自我强化、不断发展;
3. 残余风险即融合风险:即便采取了降胆固醇或抗炎单一路径治疗,心血管事件仍持续发生,这一残余风险本质上是未能打破胆固醇-炎症融合环路所致。
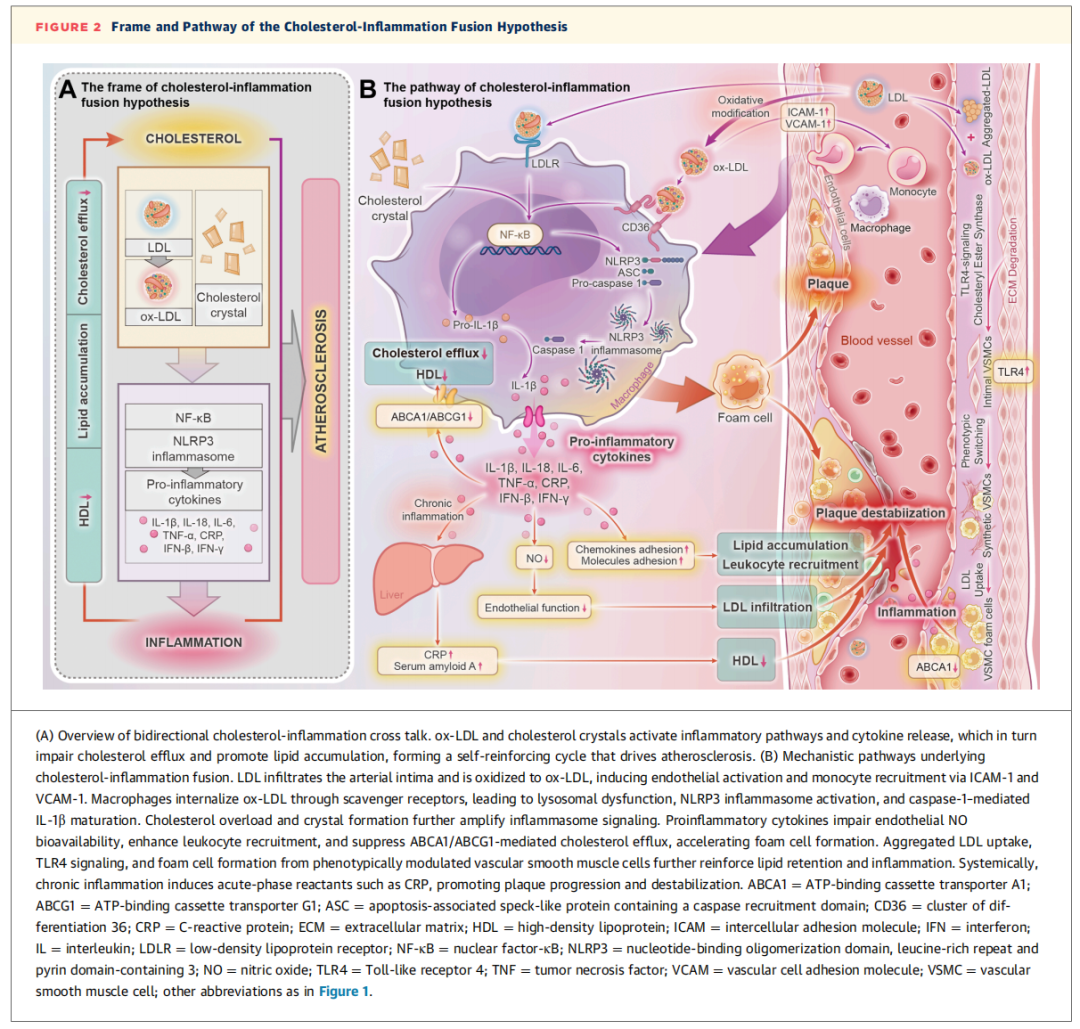
图2 胆固醇-炎症融合假说的框架和途径
机制洞悉:错综复杂的相互作用网络
机制研究揭示,胆固醇-炎症融合假说的背后,是一张由分子与细胞间相互作用构成的复杂网络。这些相互作用可归纳为三大核心融合节点:炎症小体的激活、细胞因子介导的脂质代谢紊乱,以及同时呈现脂质蓄积和促炎活性的巨噬细胞。此外,血管平滑肌细胞和内皮细胞也在其中发挥重要作用(图2B)。
胆固醇:炎症的始动因子
若将血管壁视作战场,脂质则兼具“作战者”与“挑衅者”的双重身份。当胆固醇超出其可溶性载体的结合能力时,会析出形成坚硬的针状结晶。同时,胆固醇并非仅仅是动脉粥样硬化斑块的结构组成成分,更是一种强效的炎症触发因子。当血管内膜中的胆固醇浓度超过其溶解阈值,便会形成尖锐、坚硬的结晶结构。巨噬细胞会吞噬这些胆固醇结晶,进而导致溶酶体膜破裂,组织蛋白酶释放;组织蛋白酶作为损伤相关分子模式,会激活核苷酸结合寡聚化结构域样受体蛋白3(NLRP3)炎症小体的核心调控因子。
氧化型低密度脂蛋白(ox-LDL)的作用同样关键,其不仅是血脂异常的生物标志物,还能通过清道夫受体[尤以分化簇36(CD36)为核心]主动诱导炎症小体组装,进而激活核因子-κB(NF-κB)通路,上调白介素-1β前体(pro-IL-1β)的表达。敲除CD36基因的巨噬细胞在接触ox-LDL后无法分泌白介素-1β(IL-1β),这一结果印证了CD36在该过程中的关键作用。
上述反应最终会通过半胱天冬酶-1(caspase-1)依赖的途径,促使IL-1β成熟释放;成熟的IL-1β会进一步诱导血管内皮黏附分子的表达,促进白细胞募集,并刺激白介素-6(IL-6)、肿瘤坏死因子-α(TNF-α)等下游细胞因子的分泌。综上,胆固醇结晶与ox-LDL共同作为炎症始动因子,将脂质过载与无菌性炎症直接关联,为后续自我维持的炎症循环奠定了基础(图3A)。
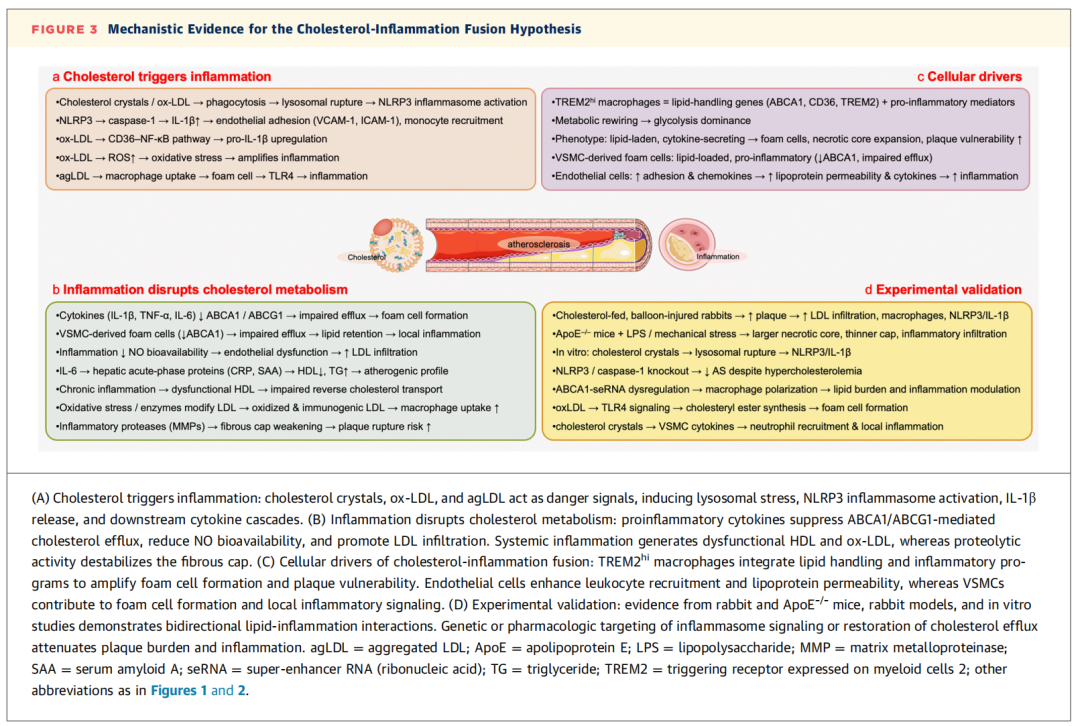
图3 胆固醇-炎症融合假说的机制证据
除胆固醇结晶和ox-LDL外,聚集型低密度脂蛋白(agLDL)也参与了炎症的启动过程。agLDL由脂蛋白颗粒聚集并与基质成分结合形成,能被巨噬细胞高效摄取,促进泡沫细胞形成和细胞内胆固醇蓄积,而这两者正是动脉粥样硬化病变炎症微环境的核心特征。尽管agLDL单独诱导细胞因子产生的具体机制尚未完全阐明,但越来越多的证据表明,其在动脉粥样硬化进展中兼具脂质负荷诱导和促炎调控的双重作用。
炎症:胆固醇代谢的破坏者
炎症级联反应会反向干扰胆固醇的稳态平衡。IL-1β、TNF-α、IL-6等促炎细胞因子会抑制巨噬细胞中腺苷三磷酸结合盒转运体A1(ABCA1)和G1(ABCG1)的表达,破坏胆固醇外流过程,加速泡沫细胞的形成。血管平滑肌细胞来源的泡沫细胞占斑块中泡沫细胞总数的一半以上;且因ABCA1表达水平较低,血管平滑肌细胞的胆固醇外流能力受损,进一步加剧了脂质滞留和局部炎症信号的传递。
此外,炎症细胞因子还会降低血管内皮一氧化氮(NO)的生物利用度,破坏内皮屏障的完整性,使LDL更易侵入血管内膜。 在全身层面,IL-6会刺激肝脏合成C反应蛋白、血清淀粉样蛋白A等急性期蛋白,导致HDL水平降低、甘油三酯水平升高。慢性炎症还会改变HDL的组成结构,形成功能异常的HDL颗粒,使其无法正常介导逆向胆固醇转运。与此同时,氧化应激和酶促修饰会让LDL颗粒的免疫原性增强,进一步促进巨噬细胞对其的摄取。
炎症细胞还会加剧斑块的不稳定性:基质金属蛋白酶等蛋白水解酶会降解斑块的纤维帽,使其结构变薄弱,增加斑块破裂的风险,这一过程将炎症对胆固醇代谢的反馈作用与急性心血管临床事件直接关联。由此可见,炎症并非单纯伴随脂质沉积发生,而是会主动促进胆固醇滞留、全身血脂异常和斑块不稳定(图3B)。
胆固醇-炎症融合的细胞驱动者
单细胞核糖核酸测序(scRNA-seq)技术揭示,动脉粥样硬化病变中存在一类特殊的巨噬细胞亚群,其可同时表现出脂质蓄积和促炎活性的双重表型。髓系细胞触发受体2高表达(TREM2hi)巨噬细胞是该融合状态的典型代表:这类细胞同时富集脂质代谢相关基因(ABCA1、CD36、TREM2)和炎症介质相关基因,且代谢模式发生重编程,以糖酵解代谢为主。TREM2hi巨噬细胞集中体现了动脉粥样硬化的融合疾病特征——胆固醇外流能力受损、细胞因子分泌增强,且炎症活性持续存在。
从功能上看,这类兼具双重活性的巨噬细胞是脂质-炎症环路的“放大器”,会促进泡沫细胞形成、斑块坏死核心扩大,加剧斑块的不稳定性。除巨噬细胞外,血管平滑肌细胞来源的泡沫细胞也会增加斑块的脂质负荷,并调控局部炎症反应,这表明多种细胞类型共同参与了胆固醇-炎症的融合过程。
血管内皮细胞同样是胆固醇-炎症融合的重要参与者,可通过上调黏附分子和趋化因子的表达,调控白细胞的募集与跨内皮迁移;通过增加血管内皮对致动脉粥样硬化脂蛋白的通透性,促进脂质侵入;还能分泌促炎细胞因子,放大血管壁内的局部免疫反应。这些细胞在动脉粥样硬化斑块中大量存在,而在健康血管中则无此特征,这也印证了其作为胆固醇-炎症融合假说细胞驱动者的核心作用(图3C)。
基础研究证据:脂质与炎症的相互作用
在饲喂胆固醇并经球囊损伤血管的家兔模型中,血管损伤与高胆固醇血症产生协同作用,加速斑块形成,具体表现为低密度脂蛋白浸润增加、巨噬细胞募集增多,同时 NLRP3 炎症小体激活,IL-1β 水平升高。在载脂蛋白 E 基因敲除小鼠模型中,额外的炎症刺激会进一步加剧斑块的体积增大与结构复杂化,表现出薄纤维帽、大坏死核心等人类易损斑块的典型特征。
体外实验中,巨噬细胞接触胆固醇结晶后会发生溶酶体破裂,进而触发炎症小体组装与IL-1β 的释放。即便小鼠仍处于高胆固醇血症状态,敲除 NLRP3 或半胱天冬酶 - 1 基因也能显著减轻动脉粥样硬化病变程度。研究还发现 ABCA1 超级增强子 RNA 等新型调控因子可调控巨噬细胞极化与胆固醇稳态,在载脂蛋白 E 基因敲除小鼠中过表达该因子,能降低小鼠体内脂质负荷与全身细胞因子水平,同时减小斑块体积(图3D)。
除了以巨噬细胞为核心的作用机制,血管平滑肌细胞也参与了动脉粥样硬化中脂质与炎症的相互作用过程。在人类动脉粥样硬化早期病变中,脂质浸润会促使血管平滑肌细胞发生表型转换,向泡沫细胞与巨噬细胞样细胞转变,且脂质负荷的增加与病变进展、炎症特征呈正相关。此外,ox-LDL可通过激活血管平滑肌细胞中的 Toll 样受体 4 介导的炎症信号通路,上调胆固醇酯合成相关关键酶的表达,进而独立于巨噬细胞促进泡沫细胞形成。部分体外模型还发现,接触胆固醇结晶的血管平滑肌细胞会通过释放炎症介质调控中性粒细胞的募集与活化,从而影响局部免疫反应。
这一机制框架为胆固醇-炎症融合假说奠定了坚实的生物学基础。
临床证据:双通路验证
胆固醇-炎症融合假说指出,单独靶向胆固醇或炎症通路均无法完全消除心血管疾病风险。临床试验证据也一致印证了单药治疗的局限性,同时证实了双通路调控的获益优势(表 1、图 4)。
表 1 多项具有里程碑意义的临床试验强调剩余风险并验证胆固醇-炎症融合假说
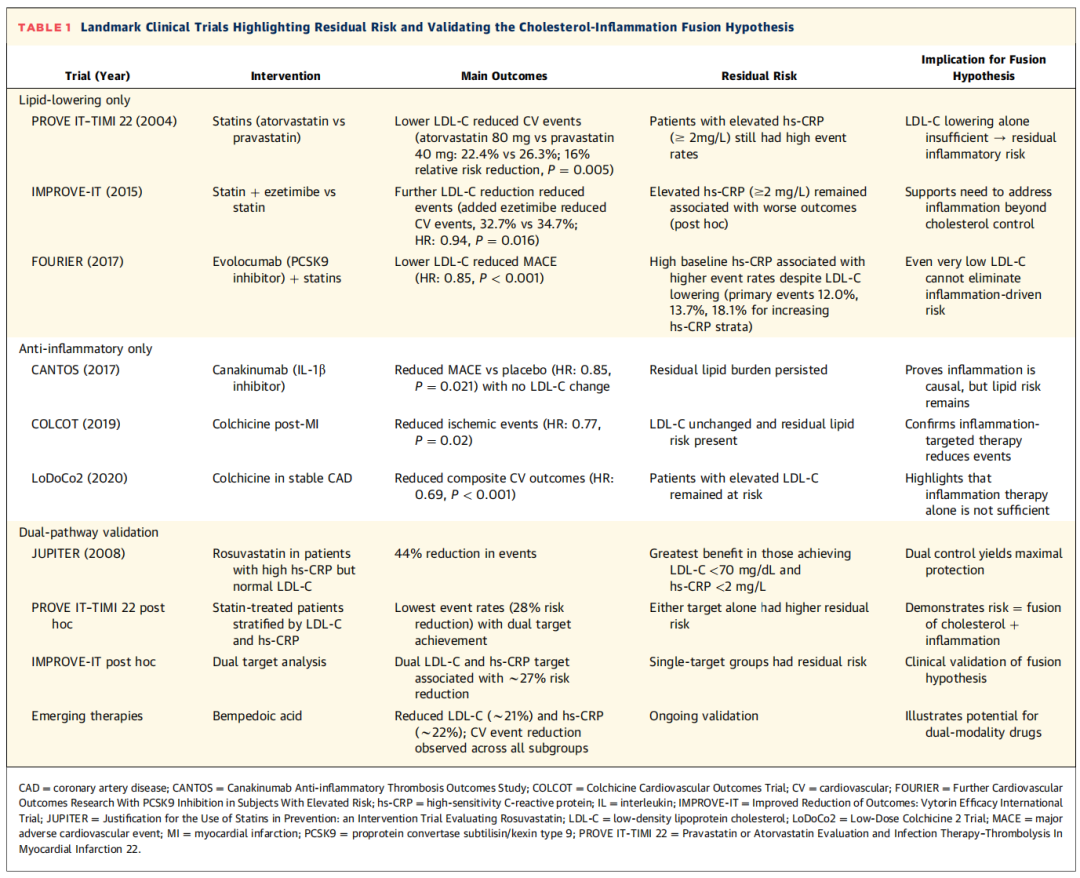
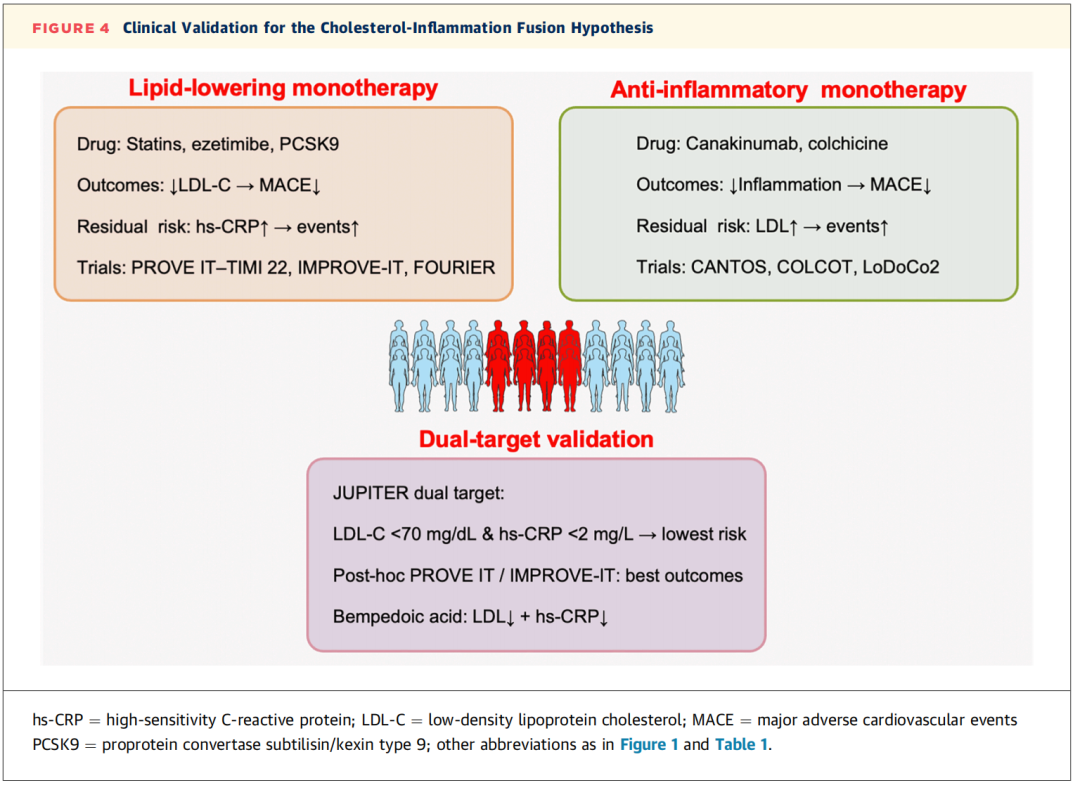
图 4 胆固醇-炎症融合假说的临床验证
降脂治疗:有效但存在残余炎症风险
他汀类、依折麦布、前蛋白转化酶枯草溶菌素 9(PCSK9)抑制剂等降脂药物能显著降低LDL-C水平,已成为动脉粥样硬化性心血管疾病预防的基石药物。然而,即便如此,患者仍存在显著的心血管残余风险。多项研究结果表明,单纯降脂无法消除心血管疾病风险,同时也凸显了炎症是引发残余风险的关键因素。
抗炎治疗:有效但存在残余脂质风险
CANTOS 试验首次证实了炎症的互补作用:采用卡那单抗选择性抑制IL-1β可在不影响LDL-C水平的前提下,降低复发性主要不良心血管事件的发生风险。秋水仙碱这类作用范围更广的抗炎药物,后续也证实了其心血管保护获益。但此类抗炎治疗并不能替代降脂治疗,患者仍存在显著的残余风险。
双通路验证:脂质与炎症调控的协同获益
对胆固醇-炎症融合假说最有力的验证,来自同时评估脂质与炎症通路的相关研究。这些研究结果具有重要的转化意义:心血管残余风险并非单纯的脂质风险或炎症风险,而是二者融合形成的融合风险。兼具脂质与炎症双调控活性的药物也成为该理论范式的新兴代表,例如苯丁酸瑞舒伐他汀(苯丁酸即贝派地酸,Bempedoic acid)不仅能降低低密度脂蛋白胆固醇,还可降低高敏 C 反应蛋白水平,实现对脂质与炎症通路的同步调控。此外,针对代谢-免疫交互作用的靶向药物的相关研究正在开展,这类药物或能进一步优化双靶点干预策略。
综上,临床试验证据均指向一个核心结论:要实现最佳的心血管风险降低效果,需同时阻断脂质驱动与炎症驱动的病理过程。单独靶向任一通路均会遗留未被控制的残余风险,而双通路调控能带来最大的保护获益。这些研究结果直接验证了胆固醇-炎症融合假说,也为整合脂质与炎症生物标志物、制定精准治疗策略提供了充分的理论依据。
讨论
动脉粥样硬化由胆固醇代谢紊乱与血管炎症之间双向、自我放大的相互作用驱动(图5)。该模型摒弃了“血脂异常与血管炎症相互独立、并行作用”的传统观点,强调二者的相互放大效应。
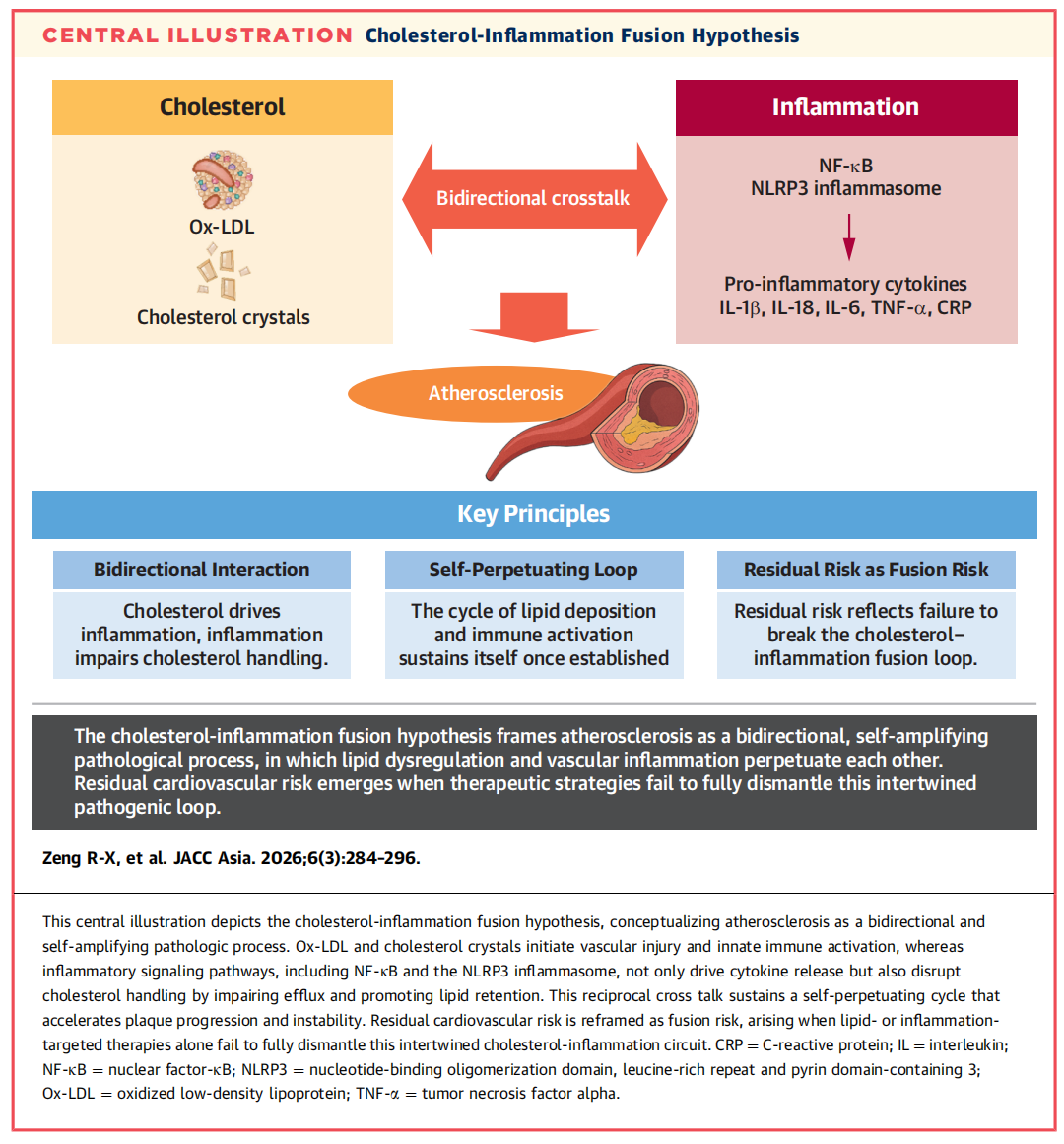
图5 胆固醇-炎症融合假说
重新定义残余风险为融合风险
残余风险并非单纯的脂质控制不足或炎症抑制不彻底,而是融合风险——由胆固醇与炎症二者的自我维持性相互作用所介导的风险。这一观点也重塑了临床决策逻辑:无论患者的低密度脂蛋白胆固醇水平或炎症生物标志物水平如何,若未能同时阻断脂质与炎症的相互作用环路,患者仍会面临心血管事件风险。
转化应用意义
将心血管残余风险重新定义为脂质负荷与血管炎症共同驱动的融合风险,这一理论框架对临床诊疗具有直接的指导意义。相应地,心血管风险分层也应从传统的胆固醇中心模型,向脂质-炎症整合评估模型转变。在近期临床实践中,可基于指南推荐的生物标志物开展这一评估策略:以LDL-C作为核心脂质靶点,以高敏 C 反应蛋白作为最经广泛验证的残余炎症风险指标。
值得注意的是,最新研究证据显示,界定炎症风险升高的高敏 C 反应蛋白最佳阈值存在人群差异,多项东亚人群研究证实,相较于西方人群采用的≥2 mg/L的临界值,更低的高敏 C 反应蛋白水平即具有预后价值。因此,纳入人群特异性的高敏 C 反应蛋白阈值,或能进一步优化融合风险分层与患者筛选。
除上述核心标志物外,脂蛋白(a)[Lp(a)]、残余胆固醇、IL- 6、TNF-α 等上游细胞因子在内的其他脂质组分与炎症介质,或能为风险分层与机制研究提供补充信息,但目前临床指南尚未将其列为常规治疗靶点。
在高危患者诊疗或科研场景中,影像学技术可进一步补充融合风险评估。正电子发射断层扫描等分子影像学技术可直观显示血管的炎症活性,高分辨率磁共振成像则能清晰描绘斑块的成分与脂质负荷,二者结合可实现对斑块内脂质蓄积与炎症空间共定位的精准检测。
从治疗角度而言,该理论框架支持采用协同的双通路干预策略。现有治疗方案中,他汀类或 PCSK9 抑制剂联合秋水仙碱、IL-1β 拮抗剂等方案,或能产生协同获益。除现有治疗方案外,靶向融合节点的新型药物是未来的研究热点,包括 NLRP3 炎症小体抑制剂、TREM2hi 巨噬细胞调节剂等。此外,抑制LDL聚集、稳定载脂蛋白 B100 的治疗手段,可通过减少聚集型低密度脂蛋白滞留、减轻溶酶体应激及下游炎症信号传导,进一步优化双通路干预策略。
现存挑战与争议
目前该领域仍存在诸多亟待解答的问题:
胆固醇沉积与炎症激活的因果层级关系仍需进一步明确;
患者的遗传背景、代谢合并症、肠道菌群等个体异质性因素,或会调控融合环路并影响治疗反应,相关机制尚未阐明;
安全性问题也亟待关注,长期抗炎治疗可能增加感染风险,而强效降脂治疗则引发了机体代谢适应的相关疑问。
结论
动脉粥样硬化应被视为由胆固醇代谢紊乱与慢性血管炎症相互交织驱动的融合性疾病。认识到这一本质,要求临床将心血管残余风险重新定义为融合风险,整合脂质与炎症生物标志物以实现精准风险分层,并设计能从薄弱节点打破融合环路的治疗方案。这一视角不仅深化了我们对疾病的认识,也为临床转化创造了切实可行的契机。未来的研究应聚焦于筛选融合特异性生物标志物、在单细胞分辨率下解析分子间的相互作用、研发能有效打破脂质-炎症融合环路的双靶点干预药物,或将推动下一代心血管疾病防治手段的研发,助力我们逐步实现降低全球动脉粥样硬化性心血管疾病负担的长期目标。
JACC: Asia编委会
